Cross-posted from https://wsmoses.com/blog/2018/12/18/boaz/
Lecturer: Aram Harrow
Scribes: Sinho Chewi, William S. Moses, Tasha Schoenstein, Ary Swaminathan
November 9, 2018
Outline
Sampling from thermal states was one of the first and (initially) most important uses of computers. In this blog post, we will discuss both classical and quantum Gibbs distributions, also known as thermal equilibrium states. We will then discuss Markov chains that have Gibbs distributions as stationary distributions. This leads into a discussion of the equivalence of mixing in time (i.e. the Markov chain quickly equilibrates over time) and mixing in space (i.e. sites that are far apart have small correlation). For the classical case, this equivalence is known. After discussing what is known classically, we will discuss difficulties that arise in the quantum case, including (approximate) Quantum Markov states and the equivalence of mixing in the quantum case.
Gibbs distributions
We have already learned about phase transitions in a previous blog post, but they are important, so we will review them again. The Gibbs or thermal distribution is defined as follows: Suppose that we have an energy function 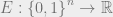

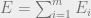
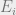

where the normalization factor in the denominator, also called the partition function, is 

In this expression, 
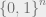
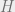
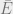
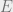
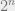
Uses of Gibbs distributions
Why is it useful to work with Gibbs distributions?
Gibbs distributions arise naturally in statistical physics systems, such as constraint satisfaction problems (CSPs), the Ising model, and spin glasses. One approach to deal with Gibbs distributions is through belief propagation (BP), which yields exact inference on tree graphical models and sometimes phase transition predictions on loopy graphs. Instead, we will focus on a different approach, namely, sampling from the Gibbs distribution.
If we want to minimize
In computer science terms, we take a sample from a high temperature because sampling is generally easier at a higher temperature than at a lower temperature. We then use that sample as the starting point for an equilibration process at a slightly lower temperature, and repeat this procedure. If we reach zero temperature, then we are sampling from the minimizers of
Gibbs distributions are used to simulate physical systems.
Gibbs distributions are used in Bayesian inference due to the Hammersley-Clifford theorem, which will be discussed next.
Gibbs distributions are also connected to multiplicative weights for linear programming (not discussed in this blog post).
Bayesian inference & the Hammersley-Clifford theorem
In order to present the Hammersley-Clifford theorem, we must first discuss Markov networks. For this part, we will generalize our setup to a finite alphabet 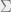

Markov chains
First, let us recall the idea of a Markov chain with variables 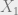
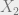


The random variables 
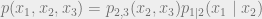
The conditional independence condition can also be written as 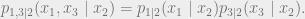
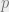
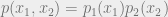

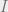
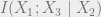

Keep in mind that conditional independence is characterized in two equivalent ways: via an algebraic condition on the distributions, and via mutual information.
Markov networks
A Markov network is like a Markov chain, but with more random variables and a more interesting structure. Imagine that we have a graph, where each node is associated with a random variable and the edges encode possible correlations. A Markov network has the property that if we take any disjoint collection of nodes 
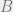
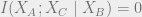

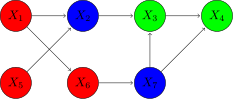
For example:
Here, if 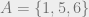
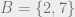
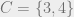
A Markov network is also called a graphical model or a Markov random field; and yet another name for them is Gibbs distribution, which is the content of the following theorem:
Theorem 1 (Hammersley-Clifford Theorem): Let 
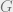

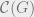
This theorem says that Markov networks are the same as Gibbs states, with the same notion of locality.
The Hammersley-Clifford theorem implies an area law for mutual information; we will explain what this is and sketch why this is true. Divide a system into two disjoint pieces 

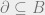

Now, we will use the fact 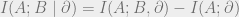

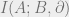

In this calculation, we have used 
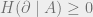
Since the mutual information only scales with the surface area of the boundary and not with the area of the two regions
Relationship to Bayesian inference
In Bayesian inference, we have a model for a system which can be very complicated. The model represents our assumptions on how parts of the system are causally related to the rest of the system. We have some observations, and we want to sample from a distribution conditionally on the fixed observations. Sampling from a conditional distribution is not the same as sampling from the original distribution, but we can still formally represent the conditional distribution as a Markov network. Therefore, sampling from Markov networks is a broadly useful task.
As an example of a complicated Bayesian model, consider a hierarchical Bayesian model [2]. Bayesian statistics requires choosing a prior distribution, and when there is a natural parameterized family of priors that a statistician can use, it may make sense to introduce a distribution over the priors; this is known as introducing a hyperparameter, and inference in the resulting hierarchical model (including computation of the posterior distribution) is frequently intractable. However, it is still desirable to work with these models because they are often more accurate than models in which the prior is handpicked by a statistician.
Sampling from Gibbs distributions
The task of sampling from an arbitrary Gibbs distribution is MA-complete [3], and it is not hard to see that at low enough temperatures this problem is at least NP-hard. So, how do we sample from these distributions?
This section will discuss Monte Carlo Markov chain (MCMC) methods, namely the Metropolis-Hastings algorithm and Glauber dynamics. Readers familiar with these methods may wish to skip to the discussion of mixing in time. For readers who wish to build more intuition about Markov chains before proceeding, see the Appendix, where the simple example of the random walk on a cycle is treated in detail.
Monte Carlo Markov chain (MCMC) methods
The general approach is to use a Markov chain. Let 


The transition probabilities of the Markov chain are1 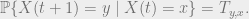


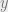

Suppose we start at a state 


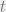

It is worth mentioning that if we are simulating the chain on a computer and we are manipulating
The justification for our algorithms is the following theorem.
Theorem 2 (Perron-Frobenius Theorem): If 


The theorem implies that 
Now, the question becomes: how does one come up with Markov chains that give you the desired stationary distribution?
Metropolis-Hastings algorithm
The first algorithm we will introduce is the Metropolis-Hastings algorithm. One more desirable feature of a Markov chain is that it satisfies detailed balance, which says 
For a Markov chain in equilibrium, the total amount of probability flowing out of
Mathematically, detailed balance implies that 
Look at the trial move 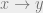
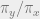

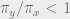

To get an idea for how the algorithm works, suppose that our underlying graph is 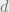

Claim: 
How does it work for a Gibbs distribution 

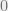

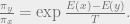
Suppose that the energy is a sum of local terms, and the underlying graph corresponds to modifying one site at at a time. What this means is that the graph is 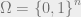
How do we choose the underlying graph? The key idea is that we do not want the majority of our moves to be rejected. A good example to keep in mind is the Ising model, where the configurations are 
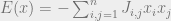

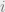
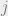
Assume that the bits are laid out in a square and 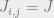


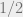

In the disordered phase, when the spins do not need to align so closely, the Metropolis-Hastings algorithm will work well. In the ordered phase, the algorithm is doomed. Indeed, suppose that most of the spins are
Glauber dynamics
This next method (Glauber dynamics) is essentially the same as Metropolis-Hastings, but this is not immediately obvious. We are at a state 


It is not obvious that these conditional distributions can be computed efficiently, but it is possible since normalizing the conditional distribution only requires summing over the possible configurations for a single random variable. On a Markov network, the conditional probability is 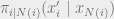

For example, in the Ising model, suppose we are at state 
![i \in [n]](https://s0.wp.com/latex.php?latex=i+%5Cin+%5Bn%5D&bg=eeeeee&fg=666666&s=0&c=20201002)

Mixing in time
Mixing in time means that the dynamics will equilibrate rapidly. It turns out that this is equivalent to mixing in space, which means that
People have known about the Metropolis-Hastings algorithm since the 1950s, but only recently have researchers been able to prove convergence guarantees for the 2D Ising model. There is a large gap between theory and practice, but in some situations we can prove that the algorithm works.
Sampling from the distribution is roughly equivalent to estimating the partition function (sampling-counting equivalence). There have been many papers addressing tasks such as estimating the non-negative permanent, the number of colorings of a graph, etc.2 A dominant way of accomplishing these tasks is proving that the Metropolis-Hastings algorithm converges for these problems. It is easy to find algorithms for these problems that converge to Gibbs distributions, but the convergence may take exponential time.
We will look at the situation when the energy function looks like the Ising model, in the sense that the interactions are local and reflect the structure of some underlying space. Also, assume that the interactions are of size 
- High temperature regime: The system is very disordered, and in the limit as the temperature approaches infinity, we get the uniform distribution.
- One-dimension: In 1D, we can exactly compute the partition function using dynamic programming. Before, we mentioned that if there are a sea of
In this part of the blog post, we will try to be more proof-oriented. We will start by explaining why it is plausible that high temperature means that the chain will mix rapidly in time.
Coupling method
One method of proving rates of convergence for Markov chains is by analzying the spectral gap. Another method is the coupling method.
The idea behind the coupling method is to start with two configurations 

The key part is that there is still some freedom with respect to what the dynamics looks like. In particular, we are allowed to correlate the 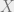



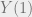


Assume we have some sort of distance function 




Initially, the two particles can be 
The distance between probability distributions is defined as follows. Let 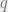

In this expression, 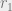




It suffices to consider when
Theorem 3: Let 




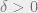
![\;\mathrm{\mathbb E}\bigl[d\bigl(X(1),Y(1)\bigr) \bigm\vert \bigl(X(0),Y(0)\bigr) = (C,C')\bigr] \le (1-\delta)d(C,C')](https://s0.wp.com/latex.php?latex=%5C%3B%5Cmathrm%7B%5Cmathbb+E%7D%5Cbigl%5Bd%5Cbigl%28X%281%29%2CY%281%29%5Cbigr%29+%5Cbigm%5Cvert+%5Cbigl%28X%280%29%2CY%280%29%5Cbigr%29+%3D+%28C%2CC%27%29%5Cbigr%5D+%5Cle+%281-%5Cdelta%29d%28C%2CC%27%29&bg=eeeeee&fg=666666&s=0&c=20201002)
for all neighboring pairs

Glauber dynamics at high temperature
Recall that in Glauber dynamics, we pick a site
- Pick a random
- If
, then set
(if the neighborhoods of the two points agree, then update them the same way). Otherwise, update them using the best possible coupling, i.e., pick a coupling for
which minimizes
.
So if 
Assume that the degree of the graph is 



(with probability
): Nothing changes;
(with probability
): We picked the one spot in which the two configurations differ. The neighborhoods of
(with probability
): We could have different updates. Here, we have to use the high temperature assumption, which says that if we change one bit, the probability of a configuration cannot change too much.In the Ising model,
. Changing
, so the expected distance afterwards is
.
Adding these cases up to get the overall expected distance gives
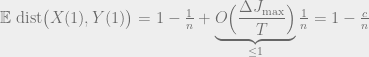
for 
Temporal and spatial mixing equivalence
The analysis of Glauber dynamics at high temperature is already a version of the equivalence between mixing in time and mixing in space. It says that if the correlations even with the immediate neighbors of a node are weak, then Glauber dynamics rapidly mixes.
Now, we want to consider the situation in which there can be strong correlations between immediate neighbors, but weak correlation with far away sites. We want to show that spatial mixing implies temporal mixing.
We will give a few definitions of correlation decay. (Note: The definitions of correlation decay below are not exactly the ones from Aram’s lecture. These definitions are from [5] and [6].)
For non-empty 



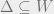



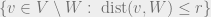
- (Weak decay of correlations) Weak spatial mixing holds for
such that for any subset
- (Strong decay of correlations) Strong spatial mixing holds for
such that for every
differing only at site
,
- (Strong decay of correlations) Strong spatial mixing in the truncated sense holds for
if there exist
such that for all functions
which depend only on the sites at
and
respectively and such that
,
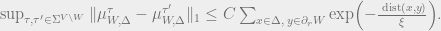
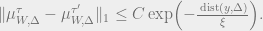

Here, 

It is a very non-obvious fact that all of these notions of spatial mixing are equivalent. We will sketch a proof that strong correlation decay implies that 
The idea is to use another coupling argument. Let 

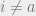


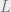
If 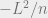

Quantum systems
The quantum version of Markov chains has many more difficulties. The first difficulty is that the Hammersley-Clifford theorem (which we have been relying on throughout this blog post) fails.
Notation
To properly discuss what we mean, let’s set up some notation. Readers already familiar with density matrices, quantum entropy, and quantum mutual information may wish to skip to the next subsection. Most of the time we discuss quantum objects here, we’ll be using density matricies, often denoted 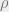

A density matrix is a positive semidefinite matrix with trace 
For example, we can consider a situation in which there is a 

Density matricies are generally useful for a lot of tasks, but for our purposes a density matrix will be used to discuss both the classical and quantum “uncertainty” we have about what state we have.
Now let’s also talk about a second important piece of notation: the tensor product. Often when discussing quantum states, it is important to discuss multiple quantum states simultaneously. For example, Alice has one system
For instance, let us consider the following state:

This particular state has the property that Alice and Bob will always both measure 

These are all notations for the same state. Let’s now talk about this state in the context of a density matrix. The density matrix of this state is as follows:
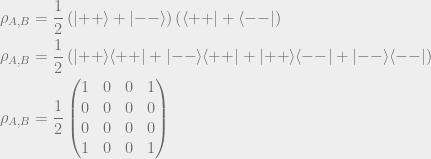
Writing the density matrix 
A crucial operation that one will often perform using density matricies is the partial trace. The partial trace is a way of allowing us to consider only a smaller part of the larger part of the system, while taking into account the influence of the larger system around it.
Here’s an example: Suppose Bob wants to know what his state is. However, Bob really doesn’t care about Alice’s system and just wants to know what the density matrix for his system is. Bob’s density matrix is simply the following density matrix (a 50% chance of being in 
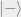

More explicitly, we could write the following:
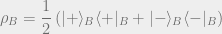
The partial trace is an operation that will let us take our original density matrix 

So how do we do this? We simply sum over the state
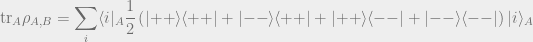
This is easier to evaluate using certain choices of notation:

This gives us the answer that we had expected.
We now have all of the tools we need to talk about quantum entropy. Intuitively, entropy can be thought of as the amount of uncertainty we have for our system, or equivalently the amount of information it takes to define our system. The entropy for a quantum system

Note that here we use the shorthand 
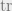

This definition intuitively makes sense since we can think of conditional entropy as the amount of information it takes to describe our joint system 
We can now discuss quantum mutual information, the amount of information that measuring system
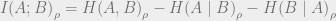
We can now finally discuss quantum mutual information (QCMI), defined as follows: 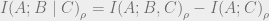

The QCMI equals
Recovery Maps
Here, the algebraic characterization is more grueling. We have

Equivalently,
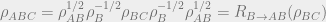
Here, 

Suppose 





However, quantum Gibbs states are not, in general, quantum Markov chains. The failure of this statement to hold is related to topological order, which is similar to the degrees of freedom that show up in error correcting codes.
Quantum Markov Networks
Here, we will formally define a quantum Markov network. The reference for this is [7].
Let 



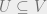




A quantum Markov network is called positive if
Now, consider the following example. First, we introduce the Pauli matrices
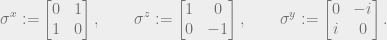
We define a Hamiltonian on three qubits

(Juxtaposition in the above expression signifies the tensor product as discussed before.) Finally, for 

The Hamiltonian here has local terms which correspond to interactions 


Important Results
We will briefly discuss the results of two papers.
- [8] This paper shows that mixing in space implies mixing in time in the quantum case. However, the result of the paper only applies to commuting Hamiltonians. For commuting Hamiltonians, it turns out that quantum Gibbs states are quantum Markov networks. They use a version of Glauber dynamics, which can be simulated on a quantum computer but are also plausible dynamics for a physical system in nature. This is a difficult paper to read, but it is worth digesting if you want to work in the field.
- [9] This second paper is much easier and more general, covering non-commuting Hamiltonians, but it requires more conditions. They give a method of preparing the Gibbs state which can run on a quantum computer, but the dynamics are not plausible as a physical system because they are too complicated. The more complicated dynamics allows them to make the proof work. The paper also uses QCMI.They have two assumptions. The first assumption looks like mixing in space (weak correlation decay). The second assumption is that the state looks approximately like a quantum Markov network (this is definitely not met in general). A very important paper in this space is a recent breakthrough ([10]) which characterizes quantum Markov chains. They show that if the QCMI is bounded by
is
-close to
, i.e., low QCMI implies that the recovery map works well. This is trivial to prove classically, but very difficult in the quantum world.The algorithm in [9] is very elegant. Essentially, we take the entire system and punch out constant-sized boxes. If we can reconstruct the region outside of the boxes, then we can use the recovery maps to reconstruct the regions inside of the boxes, and the boxes are far apart enough so they are almost independent. For this argument, we must assume that the QCMI decays exponentially. Whenever we have exponential decay, we get a correlation decay that sets the size of the boxes. It is very difficult to condition on quantum states, but recovery maps provide a sense in which it is meaningful to do so. The paper gives an efficient method of preparing Gibbs states and simulating quantum systems on quantum computers.
Additional reading
The standard treatment of information theory is [11]. This book contains definitions and properties of entropy, conditional entropy, mutual information, and conditional mutual information.
To see a treatment of the subject of Markov chains from the perspective of probability theory, see [12] or the mathematically more sophisticated counterpart [13]. An introduction to coupling can be found in [14], as well as [4] (the latter also contains an exposition to spatial mixing). The connection between Markov chain mixing and the so-called logarithmic Sobolev inequality is described in [15].
Appendix: Intuition for Markov chains
Random walk on the cycle
We have 
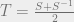
where 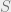


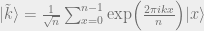
We have the same amount of amplitude at every point, but there is a varying phase which depends on 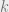


After the shift, we pick up an additional phase based on how rapidly the phase is varying. From this, we get:

The eigenvalues are

Only
How do we analyze 

If 

Spectral gap
The example of the random walk on the cycle shows that there is generally a unique stationary distribution and suggests that the speed of convergence is determined by how close the other eigenvalues are to 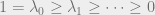
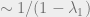
Typically, the distance of the eigenvalues from 
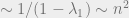
References
- S. Gharibian, Y. Huang, Z. Landau, and S. W. Shin, “Quantum Hamiltonian complexity,” Found. Trends Theor. Comput. Sci., vol. 10, no. 3, pp. front matter, 159–282, 2014.
- R. W. Keener, Theoretical statistics. Springer, New York, 2010, p. xviii+538.
- E. Crosson, D. Bacon, and K. R. Brown, “Making Classical Ground State Spin Computing Fault-Tolerant,” Physical Review E, vol. 82, no. 3, Sep. 2010.
- C. Moore and S. Mertens, The nature of computation. Oxford University Press, Oxford, 2011, p. xviii+985.
- F. Martinelli, “Lectures on Glauber dynamics for discrete spin models,” in Lectures on probability theory and statistics (Saint-Flour, 1997), vol. 1717, Springer, Berlin, 1999, pp. 93–191.
- F. Martinelli and E. Olivieri, “Finite volume mixing conditions for lattice spin systems and exponential approach to equilibrium of Glauber dynamics,” in Cellular automata and cooperative systems (Les Houches, 1992), vol. 396, Kluwer Acad. Publ., Dordrecht, 1993, pp. 473–490.
- M. S. Leifer and D. Poulin, “Quantum graphical models and belief propagation,” Ann. Physics, vol. 323, no. 8, pp. 1899–1946, 2008.
- M. J. Kastoryano and F. G. S. L. Brandão, “Quantum Gibbs samplers: the commuting case,” Comm. Math. Phys., vol. 344, no. 3, pp. 915–957, 2016.
- F. G. S. L. Brandão and M. J. Kastoryano, “Finite correlation length implies efficient preparation of quantum thermal states,” ArXiv e-prints, Sep. 2016.
- O. Fawzi and R. Renner, “Quantum conditional mutual information and approximate Markov chains,” Comm. Math. Phys., vol. 340, no. 2, pp. 575–611, 2015.
- T. M. Cover and J. A. Thomas, Elements of information theory, Second. Wiley-Interscience [John Wiley & Sons], Hoboken, NJ, 2006, p. xxiv+748.
- R. Durrett, Essentials of stochastic processes. Springer, Cham, 2016, p. ix+275.
- R. Durrett, Probability: theory and examples, Fourth., vol. 31. Cambridge University Press, Cambridge, 2010, p. x+428.
- M. Mitzenmacher and E. Upfal, Probability and computing, Second. Cambridge University Press, Cambridge, 2017, p. xx+467.
- F. Cesi, “Quasi-factorization of the entropy and logarithmic Sobolev inequalities for Gibbs random fields,” Probab. Theory Related Fields, vol. 120, no. 4, pp. 569–584, 2001.
- This is the opposite of the probabilists’ convention, i.e., the transition probability matrix that we define here is the transpose of the one usually found in most probability theory textbooks. ↩
- As a side note, it may be a good research question to investigate to what extent quantum algorithms can be used to compute summations whose terms are possibly negative. In quantum Monte Carlo, the quantum Hamiltonian is converted to a classical energy function; this conversion always works, but sometimes you end up with complex energies, which is terrible for estimating the partition function because terms can cancel each other out. ↩
- You may recognize this as the total variation norm. ↩
- Petz wrote about quantum relative entropy in 1991, way before it was cool. ↩
